
Standpunkt zu einem Artikel in Al Jazeera (11. April 2023) zu Therapien mit CRISPR-Cas
Titelfoto:
Victoria Gray wurde 2019 als erste Patientin in den USA mit einer CRISPR-Cas-basierten Therapie gegen die Sichelzellkrankheit (SCD) behandelt. Sie nahm an einer klinischen Studie teil und wurde mit ihren eigenen „reparierten“ Blutstammzellen behandelt. Sie wurde vollständig geheilt. Vier Jahre später, im Jahr 2023, war sie der Star auf dem „International Summit on Human Genom-Editing“ Bearbeitung des menschlichen Genoms und erzählte ihre Geschichte. Interessanterweise war dies die gleiche Konferenzreihe, auf der He Jiankui 2018 die international kritisierte Geburt der gen-editierten Babys Nana und Lulu präsentierte.
Der Artikel in Al Jazeera ist gut. Es ist notwendig, die ethischen und praktischen Fragen der CRISPR-Therapien zu diskutieren, aber ich glaube, dass die Diskussion umfassender sein sollte.
Keimbahntherapie
Die Keimbahntherapie war schon vor den höchst beunruhigenden Experimenten von He Jiankui weitgehend verboten. „Meistens“ bedeutet, dass selbst der sehr strenge „Ethikrat“ in Deutschland die Keimbahntherapie nicht generell ausschließt, sondern argumentiert, dass sie in ganz bestimmten Fällen tolerierbar sein könnte. Aber ist das wirklich ein Problem? Die überwiegende Mehrheit (ca. 99 %) der genetischen Krankheiten kann durch In-vitro-Fertilisation und Embryonenauswahl (PID – Präimplantationsdiagnostik) vermieden werden. Nach den Mendelschen Genetik sind mindestens 25 % der Embryonen nicht von der Krankheit betroffen oder sie tragen das mutierte Gen gar nicht (es gibt einige sehr seltene Ausnahmen dieser Regel).
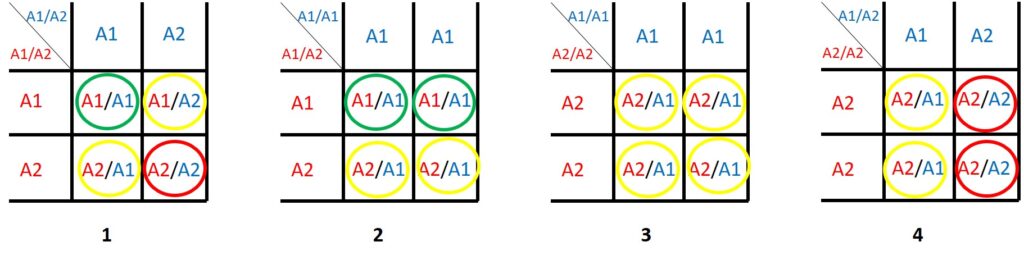
Ein bestimmtes Gen A kommt in zwei Varianten vor: die normale A1 Variante und die mutierte A2 Variante, die eine Krankheit oder Fehlfunktion verursacht. Der Mensch hat zwei Kopien jedes Gens in jeder Zelle, so dass eine Person A1/A1 oder A1/A2 oder A2/A2 sein kann. Wenn zweimal dieselbe Version vorliegt, bezeichnet man das als homozygot (A1/A1), wenn verschiedene Versionen vorliegen als heterozygot (A1/A2).
Je nach Gen können Menschen mit A2/A2 nicht lebensfähig oder so krank sein, dass sie sich nicht fortpflanzen können. A1/A2 könnte lebensfähig sein, aber ein ernstes Gesundheitsproblem haben.
Bei anderen Genen hat nur A2/A2 ein ernstes Problem, und eine A1/A2-Person erscheint völlig gesund. Allerdings können A1/A2 die „schlechte“ Genvariante an ihre Kinder weitergeben.
Keimzellen (Ei- und Samenzellen) haben nur eine Kopie von jedem Gen. Eine Person mit A1/A2 produziert die gleiche Anzahl von Eizellen (oder Spermien) mit A1 oder A2, während eine Person mit A1/A1 nur A1-Spermien (oder Eizellen) produzieren wird.
Die Quadrate oben zeigen verschiedene mögliche Kombinationen von Eltern.
Die grünen Kreise zeigen Nachkommen an, die absolut gesund sind, die roten Kreise Nachkommen, die mit Sicherheit von der Krankheit betroffen oder gar nicht lebensfähig sind. Je nach Gen können die gelben Kreise Personen darstellen, die völlig gesund sind oder von der Krankheit betroffen sein können.
Selbst wenn beide Elternteile von der Krankheit betroffen sind (Quadrat 1), haben sie eine 25 %ige Chance, ein völlig gesundes Kind zu bekommen!
Wenn ein A2/A2-Elternteil (Quadrat 3 und 4) ausreichend gesund ist, um zu leben und sich fortzupflanzen, ist es sehr wahrscheinlich, dass sein A2/A1-Nachkomme weitgehend gesund ist.
Gene, die eine Krankheit verursachen, sind selten. Daher ist die Wahrscheinlichkeit, dass bei einem Paar beide Partner das gleiche „Krankheitsgen“ tragen, eher gering. Lebensfähige Menschen, die homozygot für eine solche Genvariante sind, sind (je nach Gen) oft behindert, unfruchtbar oder haben eine geringe Lebenserwartung. Es ist sehr selten, dass sich ein Paar findet, bei dem beide Partner homozygot für eine solche Genvariante sind und einen Kinderwunsch haben. Dies wäre einer der seltenen Fälle, bei denen keine gesunden Embryonen entstehen können.
In (fast) allen anderen Fällen können genetisch gesunde Embryonen durch PID leicht selektiert werden. Das Risiko und die Belastung für Mutter und Kind sind im Vergleich zu einer CRISPR-Therapie vernachlässigbar. Auch die Zahl der verworfenen Embryonen ist bei der PID geringer. Die Keimbahntherapie ist deshalb für die Vermeidung von menschlichem Leid nicht wirklich von Bedeutung. Sie ist nur „interessant“ für eine „Optimierung“ von Menschen, d.h. das Einpflanzen von Genen oder die Veränderung von Genen, die einen Embryo „verbessern“ und die im Genom der Eltern nicht vorhanden sind. Aber das ist eine ganz andere Geschichte und sollte nicht im Zusammenhang mit der „Ausrottung menschlicher Krankheiten“ diskutiert werden.
Somatische Gentherapie
Die somatische Therapie hingegen hat in nur wenigen Jahren atemberaubende Fortschritte gemacht. Die Kosten für die Entwicklung und die Therapie selbst sind jedoch extrem hoch. Das wissenschaftliche Know-how und die erforderliche Ausrüstung sind weltweit begrenzt. Ohne einen weiteren wesentlichen Durchbruch wird die somatische Therapie für die große Mehrheit der Patienten weder finanziell erschwinglich noch technisch machbar sein. Ironischerweise dienen diejenigen, die reich genug sind, um sich eine CRISPR-Therapie leisten zu können, als „Versuchskaninchen“ für die weniger Wohlhabenden. Dabei wird die Technologie verbessert, es werden mögliche Langzeitwirkungen erforscht und langfristig kann dies auch den Preis senken. Dabei müssten die Kosten allerdings um mehr als 90% gesenkt werden (eine somatische Gentherapie kostet zurzeit 1 – 3 Millionen Dollar).
Angesichts der sehr geringen Zahl von Patienten, die bisher mit CRISPR behandelt werden (ca. 100), ist es nicht sinnvoll, zu diesem Zeitpunkt Fragen der ethnischen Zugehörigkeit und der Gerechtigkeit zu diskutieren. Die große Mehrheit der Patienten mit Sichelzellanämie und ß-Thalassämie wurde und wird in technisch fortgeschrittenen Ländern behandelt. Die überwiegende Mehrheit wird jedoch ursprünglich aus Sub-Sahara-Afrika und dem östlichen Mittelmeerraum stammen, da beide Krankheiten in diesen Regionen weit verbreitet sind.
Im Gegensatz zu Impfstoffen können CRISPR-Therapien nicht einfach als Medikament in einer Schachtel verschickt werden. Wir brauchen eine fortschrittliche Infrastruktur und spezialisierte Wissenschaftler und Ärzte, um diese Therapien dort einzusetzen, wo sie am dringendsten benötigt werden. Leider korreliert die unzureichende medizinische Infrastruktur mit der Prävalenz der Sichelzellenkrankheit (und anderer schwerwiegender Gesundheitsprobleme!).
Den Menschen in diesen Regionen zuliebe müssen wir die medizinische Kompetenz im Allgemeinen fördern und nicht so sehr Hoffnungen auf komplexe Therapien setzen, deren allgemeine und finanzierbare Anwendung noch in weiter Ferne liegt. Es wird aus verschiedenen Gründen noch viele Jahre und viele Anstrengungen brauchen, um weltweit einen ausreichend hohen medizinischen Standard zu etablieren, um auch nur die Grundbedürfnisse der Menschen befriedigt.
Es ist zu beachten, dass die somatische Gentherapie keineswegs die „Wunderwaffe“ zur Behandlung aller Erbkrankheiten ist. Auch wenn bei Blutkrankheiten Krankheiten beachtliche Erfolge zu verzeichnen sind, sind andere erbliche Erkrankungen viel schwieriger zu behandeln.
Dies bedeutet keineswegs, dass die Bemühungen um die Weiterentwicklung der CRISPR-Therapien nachlassen sollten! Das Potenzial ist extrem hoch, und ohne weitere Forschung, klinische Versuche und zugelassene Anwendungen (egal wie teuer sie sind) werden wir keine weiteren Durchbrüche erzielen und schließlich Methoden erhalten, die für alle verfügbar sind.
Autor: Wolfgang Nellen, BioWissKomm